Surface activity of hydrated protons and other ions
Although classical electrostatics theory predicts that ions are repelled
from air-water and oil-water interfaces, experiments and computer
simulations have shown that some ions actually adsorb.
The adsorption of ions has a profound influence on the interactions
between solutes in water.
It is hard to imagine a more long-standing and fundamental controversy in physical chemistry than the one concerning the affinity of protons for hydrophobic surfaces, such as oil droplets and air bubbles. Ever since the first electrokinetic measurements in the mid 19th century and the surface tensions measurements in the late 19th century, the discrepancy between the positive surface excess of protons (evidenced by a decreasing surface tension with rising acid concentration) and the negative zeta-potential of the clean air-water interface (evidenced by kinetic experiments) has stirred heated debate. In over 100 years of mounting evidence on both sides of the divide, the positions have hardened, and a fresh start from fundamental thermodynamic principles seems inevitable.
Adsorption of ions – and in particular protons – at the surfaces of colloids, membranes and biological molecules is very important for colloidal stability, biochemical interactions and nanoparticle assembly. The mechanisms and interactions responsible for the ion adsorption are subtle and immensely complex, however, and no theory has been able to capture the plethora of experimental observations so far. Apart from the intrinsic interest, for example for bubble interactions in industrial processes and atmospheric chemistry, the air-water interface represents the simplest and most fundamental case. Arguably, if we cannot understand the ion propensity at the air-water interface, we cannot hope to comprehend the more complex cases mentioned above. Computer simulations are well suited to capture the complexity of the air-water interface. Previous molecular dynamics simulations, however, have been based on heuristic force fields, and give rise to widely varying results, depending on the approach taken. Also the results of ab-initio simulations differ drastically, depending on the chosen density functional.
It is hard to imagine a more long-standing and fundamental controversy in physical chemistry than the one concerning the affinity of protons for hydrophobic surfaces, such as oil droplets and air bubbles. Ever since the first electrokinetic measurements in the mid 19th century and the surface tensions measurements in the late 19th century, the discrepancy between the positive surface excess of protons (evidenced by a decreasing surface tension with rising acid concentration) and the negative zeta-potential of the clean air-water interface (evidenced by kinetic experiments) has stirred heated debate. In over 100 years of mounting evidence on both sides of the divide, the positions have hardened, and a fresh start from fundamental thermodynamic principles seems inevitable.
Adsorption of ions – and in particular protons – at the surfaces of colloids, membranes and biological molecules is very important for colloidal stability, biochemical interactions and nanoparticle assembly. The mechanisms and interactions responsible for the ion adsorption are subtle and immensely complex, however, and no theory has been able to capture the plethora of experimental observations so far. Apart from the intrinsic interest, for example for bubble interactions in industrial processes and atmospheric chemistry, the air-water interface represents the simplest and most fundamental case. Arguably, if we cannot understand the ion propensity at the air-water interface, we cannot hope to comprehend the more complex cases mentioned above. Computer simulations are well suited to capture the complexity of the air-water interface. Previous molecular dynamics simulations, however, have been based on heuristic force fields, and give rise to widely varying results, depending on the approach taken. Also the results of ab-initio simulations differ drastically, depending on the chosen density functional.
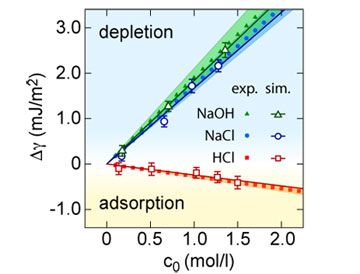
Fig. 1. Change of the air-water surface tension Δγ as a function of the bulk salt concentration c0,
in experiments and classical simulations.
The solid symbols correspond to experimental data, with the shaded area
indicating the 90% confidence interval.
The open symbols correspond to the results from classical molecular
dynamics simulations, and the solid lines are linear fits to the
simulation data.
In our work, we take a different approach.
We have optimized the molecular dynamics force fields for hydronium and
hydroxide to reproduce the macroscopic bulk solvation free energies and
the activity coefficients of the acid and base solutions [1].
This is a well-defined and controllable method to find the molecular
interaction potentials that are consistent with the measured bulk
thermodynamic quantities.
Using these force fields, we reproduce the experimental surface tension
data with remarkable quantitative accuracy (Fig. 1) [2].
The simulations clearly show a net positive excess of protons at the
air-water interface, caused by the orientation of hydronium ions in the
interfacial water layer.
We independently verify the molecular orientations using ab initio
simulations.
Apart from the high-concentration simulations, we also calculate the
potentials of mean force at vanishing ion concentration, which confirm
the conclusion that protons adsorb at the air-water interface also at
neutral pH.
| [1] | D. J. Bonthuis, S. I. Mamatkulov and R. R. Netz; Optimization of classical nonpolarizable force fields for OH- and H3O+; J. Chem. Phys. 144, 104503 (2016). | |
| [2] | S. I. Mamatkulov, C. Allolio, R. R. Netz and D. J. Bonthuis; Orientation-induced adsorption of the hydrated proton at the air-water interface; Angewandte Chemie Int. Ed., (2017). | |