Start |
Research
|
News
|
People |
Publications
|
Resources
|
Research |
Our research spans over applications of molecular quantum mechanics, computational chemistry and materials simulation to a broad selection of problems (molecular magnetism, NMR quantum computing, cluster physics, catalysis, gas separation and storage, low-dimensional materials, chiral separation, molecular sieving, interaction of light and matter), fundamental concepts of molecular spectroscopy, and method development with a special focus on machine learning implementations. Currently, we are particularly interested in the following topics:
Vibrationally induced molecular magnetismOur most recent research activities are centered around a nationally funded project named ViMoMag. Metal phthalocyanines, a highly versatile class of aromatic, planar, macrocyclic molecules with a chelated central metal ion, are topical objects of ongoing research and particularly interesting due to their magnetic properties. However, while current focus lies almost exclusively on spin-Zeeman-related effects, the high symmetry of the molecule and its circular shape suggests the exploitation of light-induced excitation of twofold degenerate vibrational states in order to generate, switch and manipulate magnetic fields at the nanoscale. The key element of this remarkable coupling is a molecular pseudorotation that can be triggered by infrared pulses and gives rise to a quantized, controllable molecular dipole moment. Three highly challenging questions can be formulated:
The goal of ViMoMag is to deliver answers to these questions. The project bridges between microwave spectroscopy of the past and modern experiments with pulsed lasers in the femtosecond regime, between simple but effective phenomenological models such as the rotating dipole moment picture and highly advanced, modern methods of electronic structure theory. 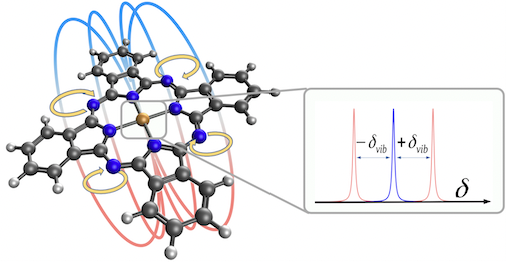 Nuclear spin control through quadrupole couplingA problem of exploiting NMR techniques for quantum computing is the inability to address a specific, single nuclear spin: Oscillating magnetic fields cannot be easily confined or screened at the nanoscale. Electric fields, on the other hand, can be efficiently routed and confined, yet they do not directly affect nuclear spins. Fortunately, there exists a remarkable coupling mechanism in certain atoms with non-spherical nuclei: Their unusual shape gives rise to a non-vanishing electric quadrupole moment, and makes them sensitive to electric fields as well. In a planned research project, we intend to study this type of interaction through quantum-mechanical simulations of suitable atomic and molecular systems. So far, we have proposed a new technology, called "Optical Nuclear Electric Resonance" or ONER, which should allow for a nanometer-resolved addressing of nuclear spins through short laser pulses. 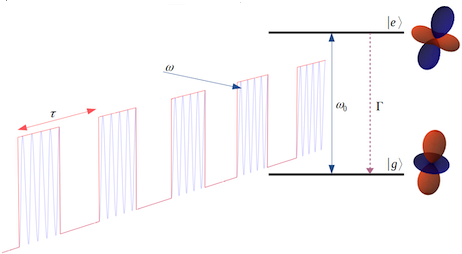 Surface chemistry, catalysis and metal cluster physicsThe properties of metals are strongly dependent on the actual particle size. Well defined nanoparticles, with atoms organized in typical crystalline facets, behave differently from monodispersed subnanometer particles with their many undercoordinated sites. Additional complexity is introduced due to differences in shape and the interaction with the support. Currently, we are investigating the intermetallic interactions in sub-nanometer sized, mixed-metallic particles on their chemical and physical properties. Particularly exciting are even smaller metallic systems, sometimes referred to as 'atomic quantum clusters'. Ongoing work with colleagus from Spain is dedicated to the photochemistry of such small, finite quantum systems, their optical features and stability with respect to temperature and oxidation.  Machine learning approaches in computational chemistryCurrently, we are undertaking several attempts to investigate the applicability of machine learning approaches to various fields of computational chemistry. Most of these investigations feed into a long term goal of our current national projects (FWF projects P 29893-N36 and PIR 8-N34), which is to perform structural studies on mixed-metallic nanoparticles, systems with sizes that make a meaningful exploration of potential energy surfaces extremely time consuming or even impossible with standard approaches. Related to this, or rather an extension of it, is the attempt to predict chemical and optical properties of metallic nanoparticles in the nanometer-range for heterogeneous catalysis, photocatalysis or optical applications. At the moment, special focus is set on the following ideas. Transition state search accelerated with machine learning algorithmsWell established methods such as nudged elastic band calculations, growing or freezing string methods and eigenvector following techniques are currently used for the calculation of reaction pathways. Yet, they all typically operate directly on the potential energy surface to be explored, which makes them rather time consuming, especially in the case of large systems with complicated high-dimensional surfaces. Two promising approaches, the application of neuronal networks and Gaussian process regression have been suggested recently. We investigate two alternative kernel-based machine learning strategies, namely support vector machine and ridged regression. A big advantage of all four algorithms in comparison to standard evaluations of the reaction pathway is the additional information gained about the local shape of the PES. Furthermore, Gaussian process regression also offers error estimates, which makes this technique particularly interesting. For details, please have a look at this JCTC article. Possible speedups for geometry optimizations in general are discussed in this JPC article. Contributions to obital-free density functional theoryOrbital-free approaches might offer a way to boost the applicability of density functional theory by orders of magnitude in system size. A crucial ingredient for this endeavor is a reasonable estimate of kinetic energy density functional. Parts of our work within FWF project P 29893-N36 is dedicated to this more fundamental research topic. On the long run, OF-DFT might solve our current problems with the description of finite metallic quantum systems. Have a look at this JCTC article. Method improvements to multi-layered feed-forward NNsBehler-Parinello-type neural networks have proven to be a useful tool for certain tasks in quantum chemistry. However, the large amount of data points needed for successful training limits the benefit over direct methods. Also, training data generation is often challenging, in particular for metallic systems with troublesome convergence behavior, and especially in regions far from from the equilibrium geometry. Therefore, we attempt to arrange these networks into a physically motivated form which enforces a correct behavior for short inter-atomic distances by construction. Acceleration of SCF algorithms via improved guessesSuitable initial guesses for the self consistent field (SCF) iteration algorithms of Hartree-Fock implementations have seen many improvements in the last decades. However, enforcing convergence e.g. for large metallic systems is still a problematic case, and automatic black box-type initial guesses often apply only to a limited selection of standard basis sets. Hückel-based methods are very effective for organic molecules, while metallic systems are preferably treated via the conceptually simple superposition of atomic densities. This is the starting point for our machine learning ansatz, which uses basis set and molecular structure information in the form of the overlap matrix S to predict the matrix elements of the density matrix. Embedded atom model potentials for metallic nanoparticles derived from NNsReliable interatomic potentials are crucial for the simulation of large systems. Machine learning approaches are typically applied to purely mathematical potential forms for the sake of generality, most notably in the atomic neural network approach suggested by Behler and Parinello. Ironically, these methods often fail at the generalization to other systems as they learn only system-specific, lacking any physical understanding such as e.g. a correct asymptotic behavior. On the other hand, physically meaningful force field models are not flexible enough to reproduce ab initio surfaces. Therefore, we aim to bridge between these two extremes by also modeling physical concepts within a neural network. Chiral separationIn this publication we suggested a new concept for the separation of chiral molecules via single-atom-thick membranes. It is based on molecular identification upon pore entrance and has the potential to exceed common techniques such as gas chromatography or high performance liquid chromatography both in terms of efficiency and cost, and might also help to reduce the environmental impact of chiral separations in the chemical and pharmaceutical industries. Currently, we are aiming at a further development of the proposed concept via a combination of electronic structure theory and molecular dynamics simulations. See also our comparison of various methods for pore propagation. 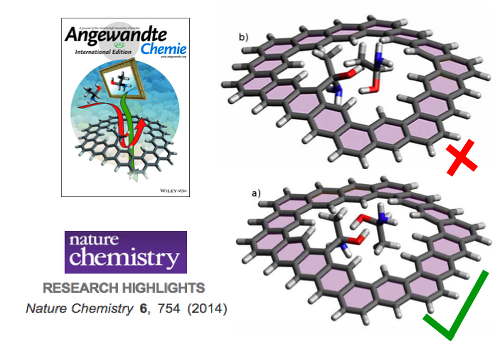 Low-dimensional materials: porous membranes and sheets of grapheneThe chemical inertness and two-dimensionality of porous graphene makes it an interesting material for applications of gas separation and storage. Its permeability for a certain gas species is mainly determined by the pore size, making it the essential parameter for separation adjustments. However, the propagation of a molecule through pores with diameters of a few angstrom needs to be treated quantum-mechanically. This became particularly obvious in our studies on the separation of helium isotopes based on counteracting effects of quantum tunneling and zero-point energy. 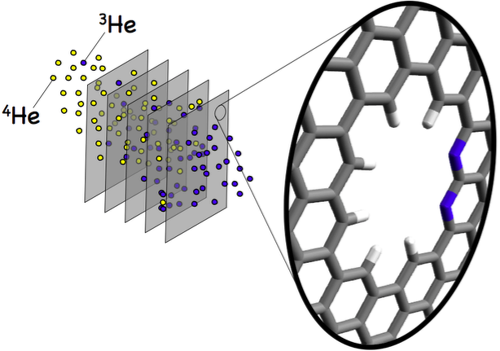 Quantum technology and ultracold chemistryIn a tight collaboration within the MOLIM framework ("Molecules in motion", a EU COST project) with María Pilar de Lara-Castells from the CSIC in Madrid we studied the effects of a superfluid helium environment on large molecules such as fullerenes and carbon nanotubes. We were interested in the effects of the helium bath on intermolecular interactions, e.g. between C60 and an alkali metal atom. Can an exothermal reaction be avoided if one dopant resides inside a He droplet while the other is "floating" on its surface? Another question we ask is how does the wetting behavior of superfluid helium differ from the classic picture, e.g. "How is the sinking an open carbon nanotube in superfluid helium different from sinking a tin can in the sea?" These questions might also be relevant in the context of quantum simulation and quantum information, where charged particles are kept floating on surfaces of liquid helium. 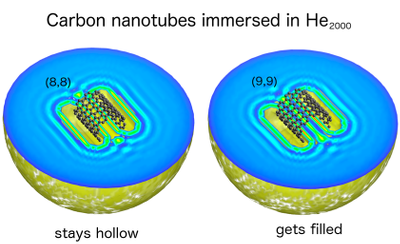 Molecular spectroscopyA good part of our research time is also spent on the assignment and interpretation of molecular spectra measured by our experimental collaborators. A big advantage of the exotic experimental technique of the Ernst group (Helium Nanodroplet Isolation Spectroscopy or HENDI) is that it gives access to spectra of rather exotic and very weakly bound species such as van de Waals-bound metal clusters in high spin states. We support their studies with ab initio calculations of ground- and electronically excited states and provide model Hamiltonian descriptions and computations for cases of spin-orbit and non-adiabatic coupling. 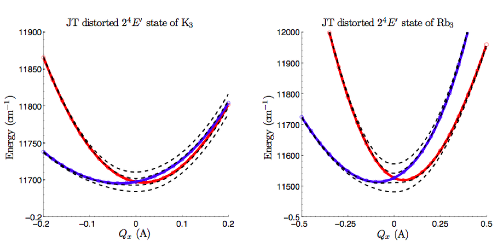  CollaborationsProf M. Arturo Lopez QuintelaProf. Lopez from the University of Santiago de Compostela is an expert on nanoparticle synthesis and a co-founder of NANOGAP, an international company with headquarters in Spain. Currently, we are investigating potential applications for smallest copper clusters, which show some potential to improve the light-harvesting abilities of titanium oxide. Prof. Em. Wolfgang E. ErnstIn a highly active in-house collaboration with the group of Wolfgang Ernst, a joint project which lasted from 2016 to 2020, we could combine local experimental skills and theory in a very effective way. Prof. Ernst is an expert on molecular beam setups in general, and the rather challenging method of heliumdroplet-mediated particle growth and deposition in particular. This technique allowed us to synthesize all kinds of mixed-metallic particles in their purest form, without solvents or templates, and to investigate their physical and chemical properties. In the past, we also supported his group with the theoretical assignment of measured laser-induced fluorescence spectra, which often involved the calculation of highly excited electronic states including non-adiabatic effects, spin-orbit coupling and in rare cases even information on hyperfine structure details. Prof. María Pilar de Lara-CastellsWith the group of María Pilar de Lara-Castells from the CSIC in Madrid we collaborate on the theoretical treatment of molecular systems with pronounced quantum phenomena, typically occuring at very low temperatures. A lot of our recent reserach focussed on molecules in a superfluid helium environment, studying the interplay of dispersion forces and quantum kinetic energy.Prof. Peter SchwerdtfegerThe collaboration with the group of Peter Schwerdtfeger in New Zealand has lasted now for several years. Originally, it focussed on the reduction of methane emissions. This gas, which is released by farm animals as well as landfills, crops and coal mines, is known to be much more effective in global warming than carbon dioxide. Our approach was to comb through the huge variety of newly developed, highly porous nanomaterials and determine the most promising candidates for a cost-effective in situ separation of CH4 from air at room conditions. We could show that porous graphene sheets with neatly adjusted pore diameters have the potential to separate methane from other gases. This discovery triggered a whole series of publications in the field of gas separation, and even led to a novel concept for the separation of bosonic from fermionic helium, a separation based on pure quantum effects! Prof. Martin Head-GordonMartin Head-Gordon, professor of theoretial chemistry at UC Berkeley, is world reknown for his contributions to method development in the field of quantum chemistry. Besides, he is also one of the founders and the scientific advisor of the Q-Chem program package. As a part of the Q-Chem developer community, we are using, debugging and extending the package, always trying to push its current limitations. A current side project of our group is the development of the Python module pyQChem, which aims at improving the usability of Q-Chem (simplified parsing, input batch file design and job programming, extensions for the calculation of thermodynamics properties). Scientifically, we were involved with recent work of the Head-Gordon group on Energy Decomposition Analayis (EDA) and its application to small metal clusters in order to study their catalytic properties. |